Czy problem zwijania białka został rozwiązany?
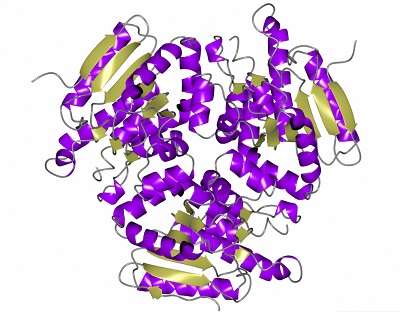
W niemal każdym wypadku (patrz poniżej na jeden wyjątek) sekwencja aminokwasów sama określa kształt wynikłego białka, bowiem prawa fizyki determinują, jak białko zwinie się, kiedy jego części składowe przyciągają lub odpychają się wzajemnie. Mogą przyjmować kształt helis, harmonijek i najrozmaitszych dziwnych zakrętów i skrętów. Tutaj jest białko, hydrataza enoilo-CoA PDB 6C7C, enzym bakterii, która powoduje wrzody skórne u człowieka. Nie jest to bardzo skomplikowany kształt, ale może być ważny w badaniu, jak spokrewniona bakteria powoduje gruźlicę, jak również do tworzenia leków przeciwko tym skórnym wrzodom:
A tutaj jest ludzka hemoglobina, stworzona z czterech łańcuchów białkowych, po dwie kopie z dwóch genów (z Wikipedii):

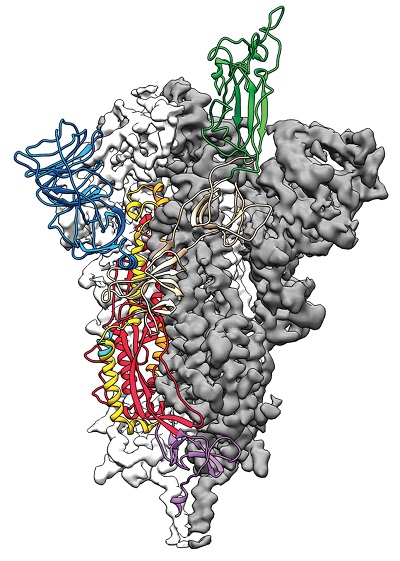
Znajomość kształtu białka jest użyteczna z wielu powodów, włącznie z powodami związanymi ze zdrowiem. Na przykład, można tworzyć lekarstwa, które wiążą się i wytrącają konkretne białka, ale jest znacznie łatwiej robić to, kiedy znasz kształt białka. (Znamy kształt tylko około jednej czwartej naszych 20 tysięcy białek.) Znając kształt białka możemy ustalić, jak patogen powoduje chorobę, np., jak “białko fuzyjne” (wypustki) wirusa COVID-19 przyczepiają się do ludzkich komórek (to pomogło w stworzeniu szczepionek). Tutaj jest wirusowe białko fuzyjne z jedną domeną wiążącą z receptorem przedstawionym jako wstążki:
Jest wiele pytań zarówno fizjologicznych, jak ewolucyjnych, które zależą od znajomości kształtów białka. Kiedy jedno białko ewoluuje w inne, jak bardzo wpływa to na jego kształt i czy zmiana kształtu może wyjaśnić zmianę funkcji? (Pamiętajmy, że w darwinowskiej ewolucji stopniowa zmiana sekwencji musi być ciągle adaptacyjna.) Jak różne kształty odorantów wchodzą w interakcje z białkami receptorów węchowych przy, w zasadzie, stosunkach jeden do jednego między kształtem białka a cząsteczką zapachu?
Jak dotąd ustalanie kształtu białka było jednym z najbardziej nużących i uciążliwych zadań w biologii. Zaczęło się dziesięciolecia temu z rentgenografią strukturalną, w której białko trzeba było krystalizować, a następnie bombardować promieniami Rentgena i pracowicie interpretować i z powrotem kalkulować oszacowania kształtów rozproszonych cząstek. (W ten sposób Franklin i Wilkins ustalili kształt DNA.) To często trwało lata dla jednego białka. Są także inne sposoby, włącznie z jądrowym rezonansem magnetycznym i nowymi metodami, takimi jak mikroskopia krioelektronowa, ale one także są niezmiernie powolne.
Teraz, w wyniku konkursu, w którym różne zespoły naukowe poproszono o użycie programów komputerowych do przewidywania struktury białek, które są już znane, ale niepublikowane, jeden z zespołów, DeepMind z Google, osiągnął zdumiewający sukces, używając sztucznej inteligencji (AI), sukces tak wielki, że inne technologie ustalania budowy białek mogą z czasem stać się przestarzałe.
Poniżej są dwa artykuły, ale w Internecie są ich dziesiątki. Pierwszy poniżej, z “Nature”, jest wyczerpujący (kliknij na link pod zrzutem z ekranu, żeby przeczytać oba):

https://www.nature.com/articles/d41586-020-03348-4
Ten artykuł, z blogu samego Deep Mind (kliknij na link pod zrzutem z ekranu), jest krótszy, ale jest tam mnóstwo użytecznych informacji, jak również pokaz, jak ściśle ich program AI przewidział budowę białka.

https://deepmind.com/blog/article/alphafold-a-solution-to-a-50-year-old-grand-challenge-in-biology
W dorocznym konkursie o nazwie CASP (Critical Assessment of Structure Prediction), poproszono sto konkurujących zespołów o odgadnięcie trójwymiarowej budowy około setki odcinków białka (“domen”). Ci, którzy z nimi pracowali, znali już budowę tych domen, ale nie była ona znana zespołom badaczy i nie była publikowana.
Metoda, jakiej użył program AI Deep Mind, by to zrobić, wykracza poza moje kompetencje, ale chodziło o “trening” programu “AlphaFold”, by przewidział budowę białek przez szkolenie programu sekwencjami aminokwasów białek, których trójwymiarowa budowa już była znana. Zaczęli parę lat temu w tym konkursie przez szkolenie programu, by przewidział odległość między każdą parą aminokwasów w białku (jeśli znasz odległości między wszystkimi parami aminokwasów, masz trójwymiarową strukturę.) W tym roku użyli bardziej wyrafinowanego programu o nazwie AlphaFold2, który, według artykułu w “Nature” “wciela dodatkowe informacje o fizycznych i geometrycznych ograniczeniach, które determinują, jak zwija się białko”. (Nie mam pojęcia, jakie są te ograniczenia; procedura jeszcze nie jest opublikowana, ale będzie na początku przyszłego roku.)
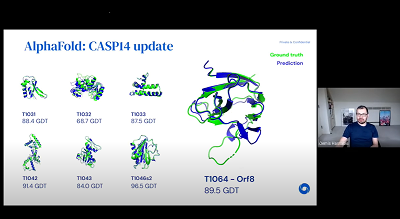
Okazuje się, że AlphaFold2 przewiduje budowę białka z niezwykłą ścisłością – często równie dobrze jak bardziej skomplikowane metody laboratoryjne, które zabierają miesiące – i robi to w parę godzin bez żadnych kosztów laboratoryjnych! W rzeczywistości, dokładność przewidzianych kształtów była około 1,6 angstrema – czyli mniej więcej szerokość jednego atomu! AlphaFold2 przewidział także kształt czterech domen białkowych, których badania naukowcy jeszcze nie zakończyli. Przed konkursem w tym roku sądzono że zabierze jeszcze co najmniej dziesięć lat, zanim AI będzie można ulepszyć do punktu, w którym będzie równie dobra jak metody eksperymentalne. Zabrało to niespełna dwa lata.
Tutaj jest gif z postu DeepMind, który pokazuje, jak dokładnie DeepFold 2 przewidział dwie struktury białka. Przystawanie do siebie zielonego (eksperymentalnego) i niebieskiego (przewidzianego przez AI) kształtu jest niezwykłe.

Nie ma wielu sytuacji, w których komputer może spowodować, że cały program eksperymentalny staje się przestarzały, ale wydaje się, że to właśnie dzieje się tutaj.
Jest jeden problem z tą metodą, ale jest on niewielki. Jak powiedział mi Matthew Cobb, w kilku wypadkach sekwencja aminokwasów nie przepowiada absolutnie kształtu białka. Jak powiedział: “Czasami ta sama sekwencja aminokwasów może dać różne izoformy [kształty, które mogą zmieniać się], co może mieć bardzo złe konsekwencje – pomyśl o prionach, w których sekwencja jest ta sama, ale budowa inna”. Priony są zmieniającymi kształt białkami, które w jednym ze swoich kształtów mogą powodować śmiertelne choroby zwyrodnienia układu nerwowego, jak „choroba szalonych krów”. Na szczęście są rzadkie, ale pokazują, że stosunek jeden do jednego między sekwencją białka a kształtem białka istotnie ma wyjątki.
Tutaj jest eleganckie wideo zamieszczone przez DeepMinds, które w osiem minut wyjaśnia sprawę:
Będziemy musieli poczekać na ukazanie się artykułu, żeby zobaczyć szczegóły, ale fakt, że program komputerowy tak dobrze przewidział kształty białek oznacza, że coś robią dobrze, a my wszyscy jesteśmy beneficjentami.
Has the problem of protein folding been solved?
Why Evolution Is True, 1 grudnia 2020
Tłumaczenie: Małgorzata Koraszewska

Emerytowany profesor na wydziale ekologii i ewolucji University of Chicago, jego książka "Why Evolution is True" (Polskie wydanie: "Ewolucja jest faktem", Prószyński i Ska, 2009r.) została przełożona na kilkanaście języków, a przez Richarda Dawkinsa jest oceniana jako najlepsza książka o ewolucji. Jerry Coyne jest jednym z najlepszych na świecie specjalistów od specjacji, rozdzielania się gatunków. Jest również jednym ze znanych "nowych ateistów" i autorem książki "Faith vs Fakt". (Jest już polskie wydanie - "Wiara vs Fakty", wydawnictwo "Stapis".) Jerry Coyne jest wielkim miłośnikiem kotów i osobistym przyjacielem redaktor naczelnej.